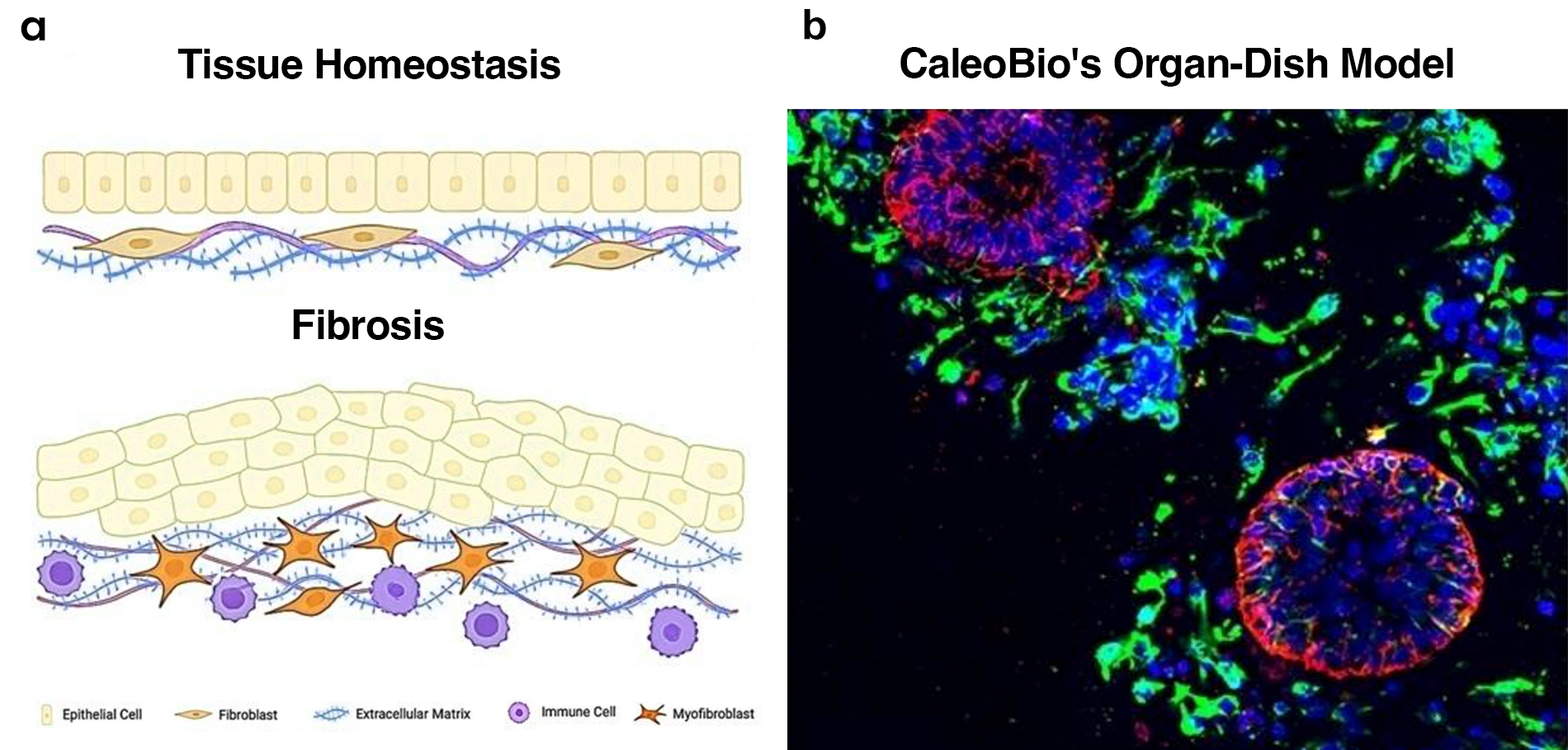
Figure 1. CaleoBio’s OD Models Support Fibrosis-like Remodeling.
(a) Schematic representation of tissue homeostasis (top) versus fibrosis (bottom), illustrating the shift from organized epithelial architecture and quiescent fibroblasts to disorganized epithelium, activated myofibroblasts, immune infiltration, and excessive ECM deposition. (b) Representative immunofluorescence (IF) image of CaleoBio’s OD model showing self-organized epithelial structures (red), embedded within a cellular microenvironment (green). Nuclei in all IF images are counterstained with DAPI (blue). scale bar = 25 μm.
Why Current Preclinical Models Fall Short in Fibrosis Research
Most preclinical fibrosis models—whether epithelial organoids, co-culture systems, or animal studies—fail to capture the cellular complexity and dynamic crosstalk that drive disease progression. Conventional epithelial-only organoids lack a functional stroma and exclude key players like mesenchymal and immune cells [7]. As a result, they miss critical mechanisms of fibrosis such as fibroblast activation, immune signaling, and ECM remodeling.
While co-culture systems attempt to reintroduce complexity, they’re often limited by poor cell integration, artificial spatial organization, and histocompatibility mismatches [8]. These systems rarely reflect the native tissue architecture or the feedback loops that exist in-vivo.
Table 1. Benchmarking CaleoBio’s OD Technology Against Existing Preclinical Models
Table 1. Benchmarking CaleoBio’s OD Technology Against Existing Preclinical Models
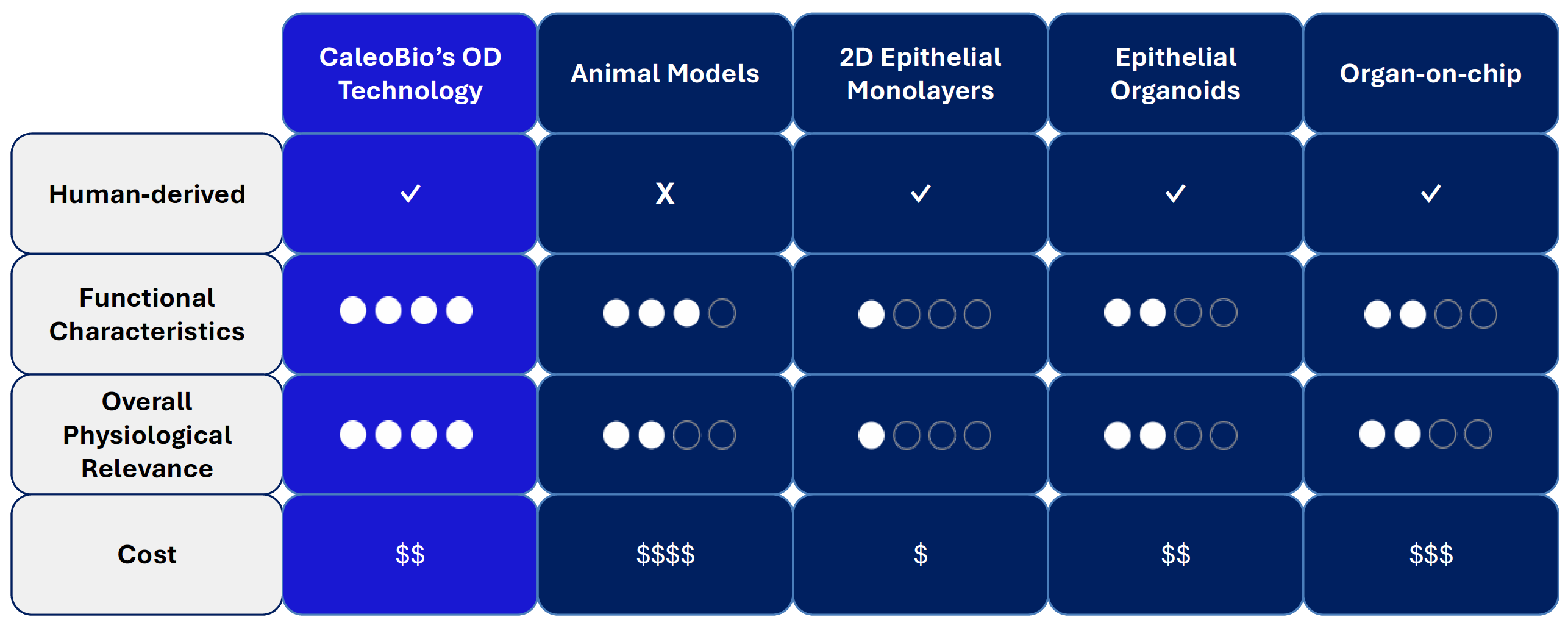
Animal models, though more complete, present major translational barriers due to species-specific differences in immune response, stromal behavior, and drug metabolism. The result is a poor correlation with human outcomes, and a major reason why fibrosis therapies continue to fail in clinical trials [9].
CaleoBio’s Organ-Dish (OD) platform directly addresses these limitations, while maintaining moderate cost. Built from patient-derived cells, OD models are self-organizing, 3D systems that naturally incorporate epithelial, mesenchymal (fibroblast/myofibroblast), and immune compartments without manual seeding. This structure allows for authentic cell–cell and cell–matrix interactions, including the feedback loops that drive tissue remodeling, inflammation, and scarring. By closing the gap between ex-vivo experimentation and in-vivo reality, CaleoBio’s OD technology provides a scalable, human-first platform to improve disease understanding, de-risk drug development, and ultimately reduce clinical trial failures in fibrotic diseases including cancer.
CaleoBio’s Technology Captures the
Hallmarks of Fibrosis
Excessive deposition of extracellular matrix (ECM), particularly type I collagen (Col1), is a hallmark of fibrotic diseases across various organs, including the lungs, liver, kidneys, and intestines [1]. This ECM accumulation reflecting the activation of fibroblasts and myofibroblasts in fibrotic tissues disrupts normal tissue architecture, leading to stiffness and impaired function.
In idiopathic pulmonary fibrosis (IPF), for instance, fibroblasts deposit Col1 within the alveolar walls, obliterating airspaces and contributing to progressive respiratory failure [3]. Similarly, in inflammatory bowel disease (IBD), chronic inflammation stimulates mesenchymal cells to overproduce Col1 intensifying mucosal damage and fibrosis [10-12].
CaleoBio’s OD models derived from patients with IBD (IBODs), effectively emulate these fibrotic processes ex-vivo (Fig. 2). In contrast to normal control ODs, which maintained a quiescent stromal profile, IBODs exhibited expanded, activated stroma with increased expression and deposition of ECM component Col1, reflecting what is observed in IBD patient biopsies (Fig. 2).
By capturing the complexity of fibrotic tissue remodeling, including the interplay between epithelial, mesenchymal, and immune components, OD models offer a robust system for translational research. They enable high-resolution studies of disease progression and therapeutic response, advancing our understanding in fibrosis treatment.
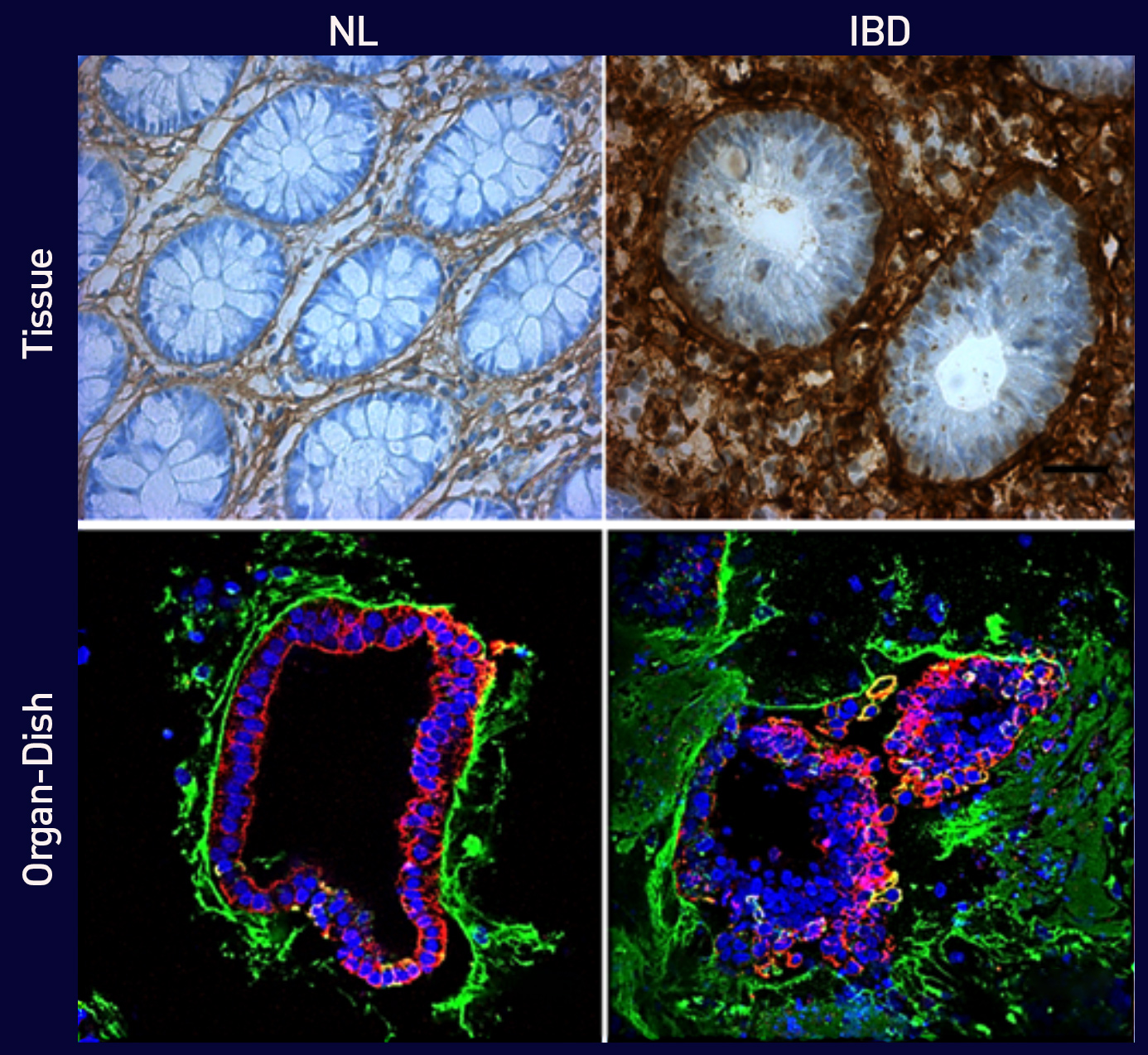
Figure 2. Excessive type I Collagen deposition in IBD vs. Normal.
(Top) Immunohistochemical staining for Col1 in primary human colonic tissue from healthy control (left) and IBD patient (right). IBD tissues show significantly higher Collagen deposition surrounding distorted crypts.
(Bottom) Immunofluorescence staining of normal control-derived (left) and IBD-derived ODs (right) showing epithelial cells (red), nuclei (blue), and Col1 (green). IBODs exhibit excessive Collagen accumulation and disrupted epithelial architecture, recapitulating the fibrotic phenotype seen in patient tissue.
Scale bars: 25 µm.
References
- Karsdal MA et al. Collagen biology and non-invasive biomarkers of liver fibrosis. Liver Int. 2020.
- Henderson NC et al. The origins and functions of myofibroblasts in organ fibrosis. Nat Med. 2013.
- Rock JR et al. Lung fibrosis and the matrix. Annu Rev Physiol. 2011.
- Meng XM et al. Fibroblast activation in organ fibrosis: biology and therapeutic targets. J Am Soc Nephrol. 2016.
- Arpaia N et al. Treg-mediated control of inflammation. Nature. 2015.
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009.
- Clevers H. Modeling development and disease with organoids. Cell. 2016.
- Kim HJ, Ingber DE. Gut-on-a-chip microenvironment induces human intestinal cells to undergo villus differentiation. Integr Biol. 2013.
- Seok J et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013.
- Heller F et al. The role of the extracellular matrix in IBD. Mucosal Immunol. 2014.
- Rieder F, Fiocchi C. Intestinal fibrosis in IBD—a dynamic, multifactorial process. Nat Rev Gastroenterol Hepatol. 2009.
- Latella G et al. Fibrosis in Crohn’s disease: mechanisms and clinical implications. World J Gastroenterol. 2011.